){kind=link}
){kind=link}
Two levels of calibration are necessary in appropriately calibrating an Ion-Selective Vibrating Probe, NVPi.
Stationary mode calibration must be performed at some level on every electrode that is used. Dynamic calibration should be done at least once for each LIX and sampling protocol. It is particularly sensitive to LIX column length, oscillation frequency, electrode shape, stepper motor speed, and interfering ions.
Stationary Calibration:
In the stationary mode, the electrode is calibrated as if it were an ordinary ion
selective electrode using methodology similar to a laboratory pH meter. One
places the electrode into a series of carefully made standard solutions of the ion
of interest and determines the mV response after the electrode has come to rest for
a suitable time. This stage should establish the range in which the
electrode gives a
linear response and how close that response complies to the Nernst Equation.
An electrode at this stage of calibration will be described as 'Nernstian'
if it has a slope [mV vs log10 concentration] which is close to the
expected (58 mV for a monovalent cation and
29 mV for a divalent cation).
For most LIXs our experience is that the slopes approach to Nernstian is more a
function of the care in preparing the standards than most other factors.
Thus, if an electrode is giving you a close but non-exactly Nernstian slope,
suspect the standards first. If systematicly non-Nernstian electrodes
are being obtained, suspect
in order, linear range of the LIX, ground electrode, electrode tip size, loss of
LIX from poorly silanized electrode glass, temperature of measurement.
You must establish confidence that your electrodes are performing like a good
pH meter measuring proton concentration. When one has such confidence,
a broken electrode can be quickly replace with a new one, applying a one-point
calibration in the physiological saline being used for the experiment.
An artificial source for dynamic calibration:
The use of a capilary tube with a finely drawn tip as an artificial source of
flowing ions was established by the developers of the technique (Kühtreiber
and Jaffe, 1990). This source provides a concentration gradient
eminating from its tip that in an
undisturbed setting
with
minimal convection is
inversely related
to the distance from the tip to the point of measurement,
approaching with slope K the background concentration, Cb, at infinite radius,
r, when 1/r = zero. To the extent this voltage/inverse-distance
curve is linear, the µV difference associated with a change in radius is
predictable, using the assumptions of linearity and the slope and intercept of
that line.
This situation provides a physically predictable model about which one can
predict the expected µV drop over a specified distance as well as a physical
model in which actual measurements could be made. From the difference between
the predicted and actual measurements one can calculate an efficiency of the
measurement process.
µV Calibration during Oscillation (dynamic calibration):
The movement of the electrode during the µV difference measurement places
special demands on the calibration process. The speed with which the LIX
in the electrode can respond to the changed concentration after the move varies
with the LIX used as well as the geometry of the electrode. In addition,
one needs to use an artificial source from which one can predict an expected
flux of the ion of interest; and on which one can determine the efficiency of the
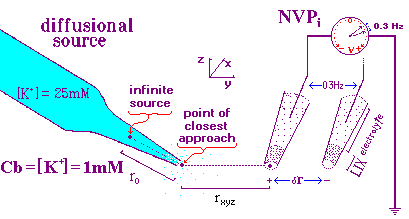
dynamic measuring process. Fig 1, below presents a cartoon of the dynamic
calibration setup.
This model was constructed for the calibration of ion probes using the 3DVIS software package in which the electrode is continuously oscillating (0.3 Hz in this example). The more advanced software, ASET, is based on a discrete movement sampling rule approach which eliminates the continuous oscillation and demands a different calibration protocol. The two protocols for 3DVIS and ASET will be explained separately. The two protocols share the following features illustrated in Fig 1.
 |
 |
eff = efficiency of the measure
S K dr r = distance to point source
dV = eff ------- / (Cb + K/r), where: dr = oscillation distance
z r2 K = slope of [ion] curve
Cb = background [ion]
S = Nernst coeficient
z = unit charge on the ion
 |
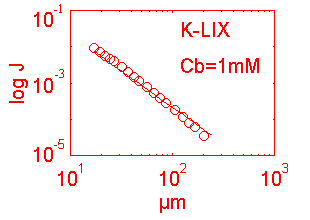 |
D = diffusion coeficient for the ion (cm-2 sec-1)
Jo = - D dC/dr , where dC = concentration differential
dr = oscillation differential
| Calibration Using 3DVIS Software | |||||||
| Ion | Do | i | Nernst slope |
LIX | t90 | Hz | eff% |
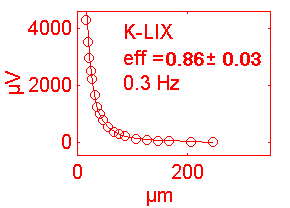
| K+ | 19.6 | +1 | 58 | K IB | <1s. | 0.3 | 70% |
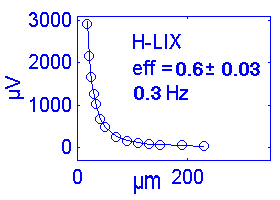
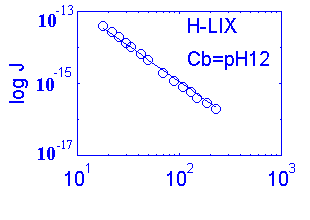
| H+ | 93.7 | +1 | 58 | H IIA | ~0.6s | 0.3 | 80% |
| Ca++ | 7.9 | +2 | 29 | Ca IIA | <5s | 0.3 | 50% |
| Mg++ | 7.1 | +2 | 29 | Mg IV | <30s | 0.25 | 30% |
| Cl- | -20.3 | -1 | -58 | ||||
| Calibration Using ASET Software | |||||||
If you have questions about calibrating the various vibrating probes or the University of Massachusetts Vibrating Probe Facility, email its PI, Joe Kunkel